Bioscience
Sequencing reveals strains behind cholera outbreak
Whole-genome sequencing of the bacterium responsible for a cholera outbreak reveals a population structure missed by traditional methods.



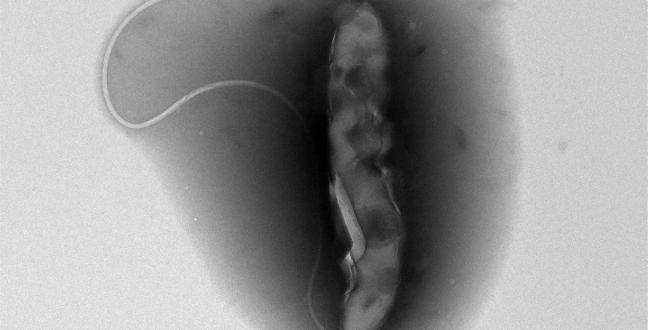

Vibrio cholerae, the bacterium responsible for the diarrheal disease.
© KAUST 2015
Deep DNA sequencing shows the 2009 cholera outbreak in the Indian city of Chandigarh was driven by two distinct strains of the pathogen — a finding which demonstrates that whole genome sequencing can improve resolution and precision over traditional molecular approaches to investigate epidemics.
Chandigarh, a city near the foothills of the Himalaya Mountains, has been struck by a number of cholera outbreaks in the past 20 years – one of these cases was during the monsoon season in 2009, when Chandigarh’s Government Medical College Hospital confirmed 61 cases of people infected with Vibrio cholerae, the bacterium responsible for the diarrheal disease.
“Whole genome sequencing allows the discovery of previously unknown variation and the emergence of new or highly diverged strains with high accuracy and speed that the traditional molecular approaches often fail to deliver,” says Arnab Pain, associate professor of Bioscience at KAUST.
Pain and his colleagues wanted to understand how the strains responsible for the outbreak compared to others around the world. They also sought to compare the resolution and accuracy of standard analytical techniques with newer methods of whole-genome sequencing. The KAUST team, in collaboration with scientists in India and the United Kingdom, characterized the genetic code of a subset of 38 clinical isolates of V. cholerae from Chandigarh1.
Using comparative genomic analyses, Pain’s team identified two strains of V. cholerae that circulated during the single 2009 outbreak in Chandigarh. These strains were similar to those found elsewhere, including in Haiti and Nepal, harboring genes for multi-drug resistance and a highly virulent form of a cholera toxin gene.
“Our results clearly indicate the prevalence of two distinct clades of V. cholerae circulating and causing disease in the same season and at the same region,” says Moataz Abd El Ghany, a postdoctoral fellow at KAUST and the first author of the research. “This has potential public health importance with respect to the prevention and control of the disease.”
Although full genome sequencing is still more expensive and less accessible than traditional approaches, it could be used by public health officials in the future as a tool to manage ongoing cholera outbreaks.
Pain says that an added benefit of sequencing-based studies is that they could help identify “more appropriate genetic markers for the development of cheap, accurate and fast diagnostic tools to subtype and discriminate between isolates.”
References
- El Ghany, M.A., Chander, J., Mutreja, A., Rashid, M., Hill-Cawthorne, G.A. et al. The population structure of Vibrio cholerae from the Chandigarh Region of Northern India. PLoS Neglected Tropical Diseases 8, e2981 (2014). | article
You might also like

Bioscience
Robust workflow built for chemical genomic screening

Bioscience
Cell atlas offers clues to how childhood leukemia takes hold

Bioscience
Hidden flexibility in plant communication revealed

Bioscience
Harnessing the unintended epigenetic side effects of genome editing
Bioscience
Mica enables simpler, sharper, and deeper single-particle tracking

Bioengineering
Cancer’s hidden sugar code opens diagnostic opportunities
Bioscience
AI speeds up human embryo model research
Bioscience