Bioscience
Stem cells reveal underpinnings of rare immune disease
Abnormal protein tied to Wiskott-Aldrich syndrome found to control gene splicing.



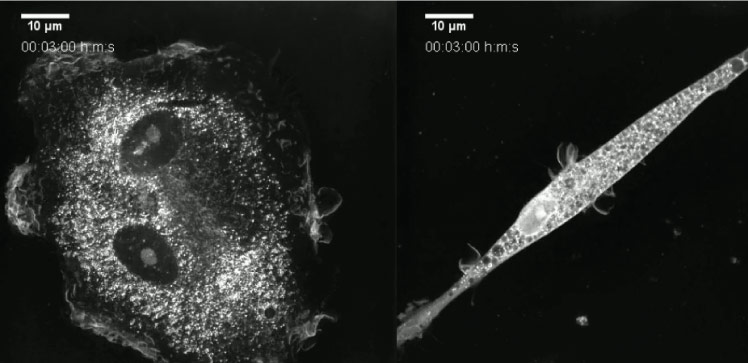
A new stem cell study by KAUST researchers helps to explain a rare genetic disease called Wiskott-Aldrich syndrome (WAS), yielding molecular clues that could lead to new treatments for a devastating immune deficiency disorder.
WAS afflicts around one in every 100,000 babies, causing frequent bleeding episodes and infections while also elevating the risk of life-threatening inflammatory diseases and certain cancers. It has been known for almost 30 years that mutations in the gene that encodes WASP — Wiskott–Aldrich syndrome protein — cause the immune abnormality, but the precise functions of this protein have long eluded scientists.
To better understand what WASP does, stem cell biologist Mo Li and his KAUST colleagues teamed up with collaborators from the Salk Institute for Biological Studies in California, USA, and created a panel of induced pluripotent stem cells (iPSCs) from patients with the immunological deficiency. They also used gene editing to either fix the mutational error of patient iPSCs or delete the entire WAS gene of a WT iPSC line, resulting in paired stem cell lines that matched in all ways except in their WAS gene sequence.
“These are powerful models that can help us understand WASP functions and the disease mechanisms of Wiskott-Aldrich syndrome in an authentic human cellular context,” says Li.
The stem cells and their differentiated progeny allowed the researchers to study the consequences of aberrant WASP activity in different immune cell lineages. They found that cells without a working version of WASP had elevated levels of another group of proteins known as RNA splicing factors, which play a critical role in processing gene transcripts so that they encode the proper recipe.
With such high levels of these splicing factors, transcripts often get truncated or end up with missing domains. General protein function suffers, and the cell becomes unwell.
In collaboration with bioengineer Samir Hamdan and his lab group, the researchers then showed that an operational version of WASP works together with a particular splicing factor, SRSF2. The team went on to show that WASP is needed to constrain the activity of SRSF2 through the production of transient liquid-like clusters of proteins and nucleic acids. Known as biomolecular condensates, these subcellular hubs normally contain WASP alongside DNA-to-RNA copying enzymes, newly synthesized gene transcripts and splicing factors including SRSF2.
“Our study reveals for the first time that WASP is a phase-separated protein that can directly participate in the process of RNA splicing,” says co-first author and Ph.D. student Baolei Yuan.
Notably, though, WASP deficiencies could be overcome by genetically targeting the activity of SRSF2. In stem cell–derived immune cells, for example, knocking down expression of this splicing factor helped prevent the release of inflammation-promoting molecules, a finding that raises tantalizing therapeutic possibilities. “SRSF2 can be a potential target for managing Wiskott-Aldrich syndrome disease,” says the other co-first author and Ph.D. student Xuan Zhou.
References
- Yuan, B., Zhou, X., Suzuki, K., Ramos-Mandujano, G., Wang, M., Tehseen, M., Cortés-Medina, L.V., Moresco, J.J., Dunn, S., Hernandez-Benitez, R. et al. Wiskott-Aldrich syndrome protein forms nuclear condensates and regulates alternative splicing. Nature Communications 13, 3646 (2022).| article
ABOUT THE AUTHOR
Baolei Yuan & Xuan Zhou
Ph.D. Students

You might also like
Applied Physics
Colorful solution to advanced disease diagnosis
Bioscience
Multi-enzyme pathway delivered into living cells
Bioscience
Brain fuel also helps wire neural connections
Bioscience
Single small-molecule model reveals insights into human embryo development

Bioscience
Can AI finally bring order to biology’s data deluge?

Bioengineering
Bio-inspired network structures for next-generation AI

Bioscience
Robust workflow built for chemical genomic screening

Bioscience