Chemistry
Anderson’s model holds for 2D materials
A simple model is shown to accurately predict the electronic properties of a combination of 2D semiconductors.




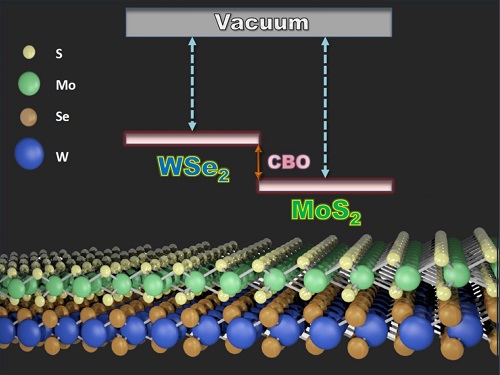
The Anderson model enables a simple way of predicting the offset of the top of the bandgap (labelled as CBO) of 2D semiconductors, such as tungsten diselenide and molybdenum disulfide.
© 2016 Ming-Hui Chiu
The defining property of a semiconductor is its so-called bandgap: the barrier that prevents electrons within a specific energy range from flowing through a material. KAUST Professor of Material Science and Engineering Lance Li and his team collaborated with colleagues from Taiwan and used a simple model to determine the band alignment in an exciting new class of semiconductors called two-dimensional transition-metal dichalcogenides (TMDs).
The simple concept of bandgap enables a single semiconducting material, such as silicon, to perform the operations required by electronic devices; however, when two or more semiconductors are combined, the device affords a broader range of functionality and boasts improved performance and efficiency. To understand how such heterostructures behave, it is crucial to know how the bandgaps of the two materials align.
Although graphene and TMDs are all atomically thin, the lack of a bandgap in graphene limits its application to electronics while the presence of a bandgap in TMDs allows them to be stacked into heterostructures. It is, however, difficult to experimentally determine band alignment between these layers because the results depend on the quality of fragile TMDs. Li and his team have now proven that the concept known as the Anderson model, a simple, computationally inexpensive way of determining band alignment, is applicable to this system1.
The Anderson model assumes that when two semiconductors are placed together, they share a common zero in their energy-band structure known as the vacuum level. Bandgap alignment can then be determined directly from calculated values of bandgaps and offsets. Until now, it was unclear whether this assumption would hold true in atomic-layer TMDs.
Li and his team addressed this by measuring the energy of the bandgap in three TMDs, molybdenum disulfide, tungsten disulfide and tungsten diselenide, using a method called ultraviolet photoelectron spectroscopy. They then applied the Anderson model to predict the band alignment. They compared these calculated values with direct experimental measurements from X-ray photoelectron spectroscopy of molybdenum-disulfide–tungsten-disulfide and molybdenum-disulfide–tungsten-diselenide heterostructures.
Agreement between the values obtained by the two methods indicated that the Anderson model holds true. The team suggests that this is because of unique van der Waals surfaces, which ensure an absence of dangling atomic bonds that would otherwise prevent the vacuum levels in the two materials from aligning.
“Our next step is to build heterojunctions based on the knowledge gained from the theory,” said Li. “We will research several heterostructures for various applications, such as solar cells and light-emitting diodes.”
References
- Chiu, M.-H., Tseng, W.-H., Tang, H.-L., Chang, Y.-H., Chen, C.-H., Hsu, W.-T., Wu, C.-I. & Li, L-J. Band alignment of 2D transition metal dichalcogenide heterojunctions. Advanced Functional Materials 27, 1503756 (2017).| article
You might also like

Chemistry
Opening a window on architectural nano-design
Bioscience
Multi-enzyme pathway delivered into living cells

Chemical Engineering
Natural solvents enable fully bio-based membranes

Chemistry
Turning infrared solar photons into hydrogen fuel

Applied Physics
Natural polymer boosts solar cells

Chemistry
Disruptive smart materials flex with real world potential
Chemistry
Catalysts provide the right pathway to green energy
Chemistry