Computer Science
AI reveals nature of RNA-protein interactions
A deep learning tool could help in structure-based drug discovery.



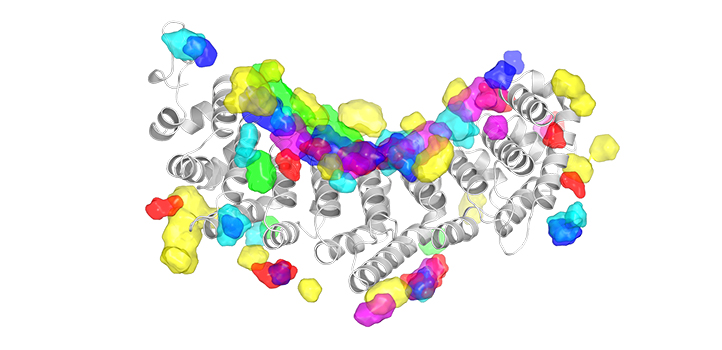
A new computational tool developed by KAUST scientists uses artificial intelligence (AI) to infer the RNA-binding properties of proteins.
The software, called NucleicNet, outperforms other algorithmic models of its kind and provides additional biological insights that could aid in drug design and development.
“RNA binding is a fundamental feature of many proteins,” says Jordy Homing Lam, a former research associate at KAUST and co-first author of the study. “Our structure-based computational framework can reveal the detailed RNA-binding properties of these proteins, which is important for characterizing the pathology of many diseases.”
Proteins routinely interface with RNA molecules as a way to control the processing and transporting of gene transcripts—and when these interactions go awry, information flow inside the cell is disrupted and disorders can arise, including cancer and neurodegenerative disease.
To better understand which parts of an RNA molecule tend to bind on different surface points of a protein, Lam and his colleagues turned to deep learning, a type of AI. Working in the laboratory of KAUST Professor Xin Gao in the Computational Bioscience Research Center, Lam and Ph.D. student Yu Li, taught NucleicNet to automatically learn the structural features that underpin interactions between proteins and RNA.
They trained the algorithm using three-dimensional structural data from 158 different protein-RNA complexes available on a public database. Pitting NucleicNet against other predictive models—all of which rely on sequence inputs rather than structural information—the KAUST team showed that the tool could most accurately detect which sites on a protein surface bound to RNA molecules or not.

This illustration depicts the training strategy and utilities of NucleicNet. RNA-protein structures in the protein data bank are stripped of their bound RNA, and surfaces of the proteins are analyzed for their physicochemical properties. The results are compiled as training input for NucleicNet. Once training is complete, the learned model can be used to predict RNA binding sites for unseen query protein structures.
© 2019 KAUST; Heno Hwang
What is more, unlike any other model, NucleicNet could predict which aspects of the RNA molecule were doing the binding, be it part of the sugar-phosphate backbone or one of the four letters of the genetic alphabet.
In collaboration with researchers in China and the United States, Lam, Li and Gao validated their algorithm on a diverse set of RNA-binding proteins, including proteins implicated in gum cancer and amyotrophic lateral sclerosis, to show that the interactions deduced by NucleicNet closely matched those revealed by experimental techniques. They reported the findings in Nature Communications.
“Structure-based features were little considered by other computational frameworks,” says Lam. “We have harnessed the power of deep learning to infer those subtle interactions.”
NucleicNet is openly available for researchers who want to predict RNA-binding sites and binding preference for any protein of interest. The software can be accessed at http://www.cbrc.kaust.edu.sa/NucleicNet/.
References
-
Lam, J.H., Li, Y., Zhu, L., Wei, J.N., Umarov, R., Jiang, H., Heliou, A., Sheong, F.K., Liu, T., Long, Y., Li, Y., Fang, L., Altman, R.B., Chen, W. Huang, X. & Gao, X. A deep learning framework to predict binding preference of RNA constituents on protein surface. Nature Communications 10, 4941 (2019).| article
ABOUT THE AUTHOR
Jordy Homing Lam and Yu Li
Alum and Ph.D. Student

You might also like
Applied Physics
Colorful solution to advanced disease diagnosis

Bioscience
Can AI finally bring order to biology’s data deluge?

Computer Science
AI-based hydrogen plant models improve power grid stability

Bioengineering
Bio-inspired network structures for next-generation AI

Computer Science
Green quantum computing takes to the skies

Computer Science
Probing the internet’s hidden middleboxes
Bioscience
AI speeds up human embryo model research

Computer Science